Population and Statistical genetics at the Bioinformatic Center at UCPH
We are meta group of labs that work with various parts of population, medical and statistical genetics at the Biocenter at University of Copenhagen. The meta group consists of 5 labs that works with focus on different organisms and systems. We apply and develop methods for analyzing large scale next generation sequencing data.
Hans Siegismund

We work on population genetics, phylogeography and speciation processes of large African mammals, mainly bovids and great apes. Another research area includes the study of evolutionary genetics of foot-and-mouth-disease (FMD) virus in East Africa.
Ida Moltke

We develop and apply statistical methods to genomic data with the purpose of gaining insights into human disease, history and evolution. For instance, by studying DNA from the Greenlandic population we recently identified a genetic variant that explains 10-15% of all cases of type 2 diabetes in Greenland. We have also looked into the migration history of the Artic and are currently investigating how the Greenlanders have genetically adapted the Arctic cold and their very fat-rich diet consisting mainly of seal and fish.
Rasmus Heller

I am interested in applying population and evolutionary genetics to answer questions about animal biology, particularly in large mammals. Most of my work has revolved around large African mammals. My research tries to address a range of topics in these species, including the historical drivers of population dynamics, how variation emerges and is retained, speciation, adaptive evolution and the relationship between phenotypic and genomic variation. I am also interested in topics of a more immediate interest in species conservation such as landscape genetics, the effect of habitat fragmentation, population connectivity etc
Anders Albrechtsen

Our group develops statistical and computational methods for analysis of genomic data including methods for performing multi-loci association studies, methods for detecting and correcting for population stratification, methods for detecting natural selection, loci dependent methods for modeling identity-by-descent and various methods for analysis of second generation sequencing data.
Software
PCAone
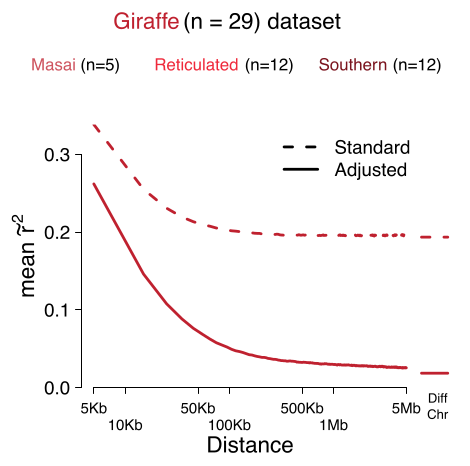
We show that population structure affect LD, pruning and clumping. We derive at new measure of LD that is corrected for population structure and admixture which improved downstream analysis
Ulises Bercovich, Malthe Sebro Rasmussen, Zilong Li, Carsten Wiuf, Anders Albrechtsen
APOH
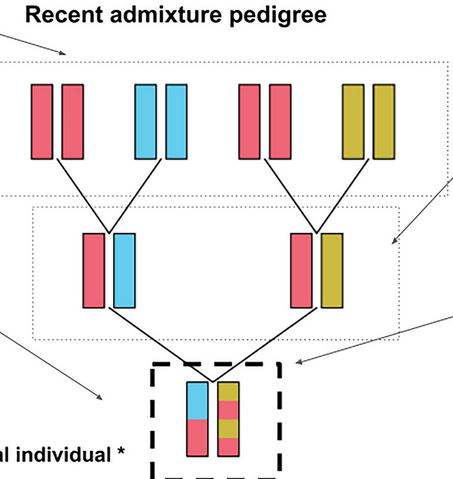
How to infer the ancestry of an admixed individuals pedigree.
Genís Garcia-Erill📨, Kristian Hanghøj, Rasmus Heller, Carsten Wiuf, Anders Albrechtsen
evalPCA
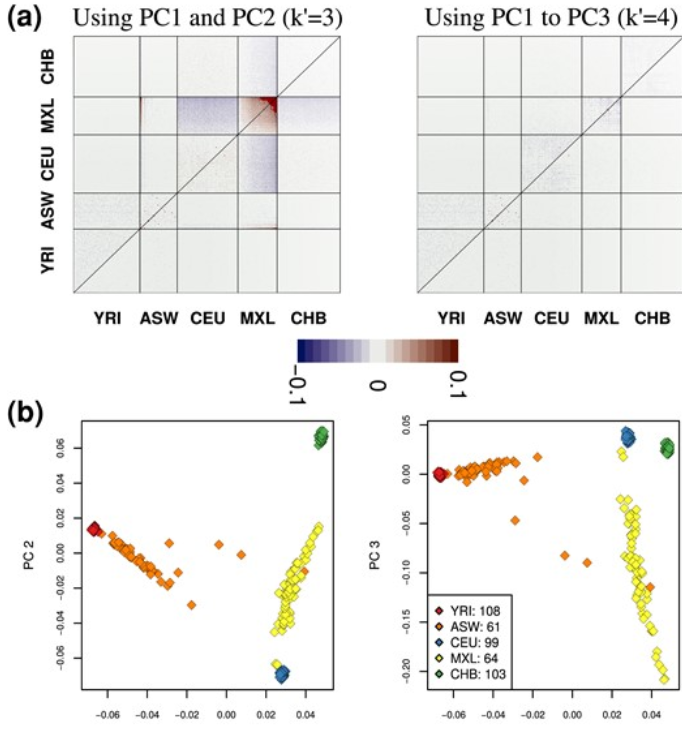
When do distances in a PCA reflex genetic ancestry? if two individuals cluster togeather does that mean they are some the same populations? Here we present a method that you can use to interpret you genetic PCA plot.
Jan van Waaij* 📨, Song Li* , Genís Garcia-Erill* , Anders Albrechtsen, Carsten Wiuf📨;
PCAone
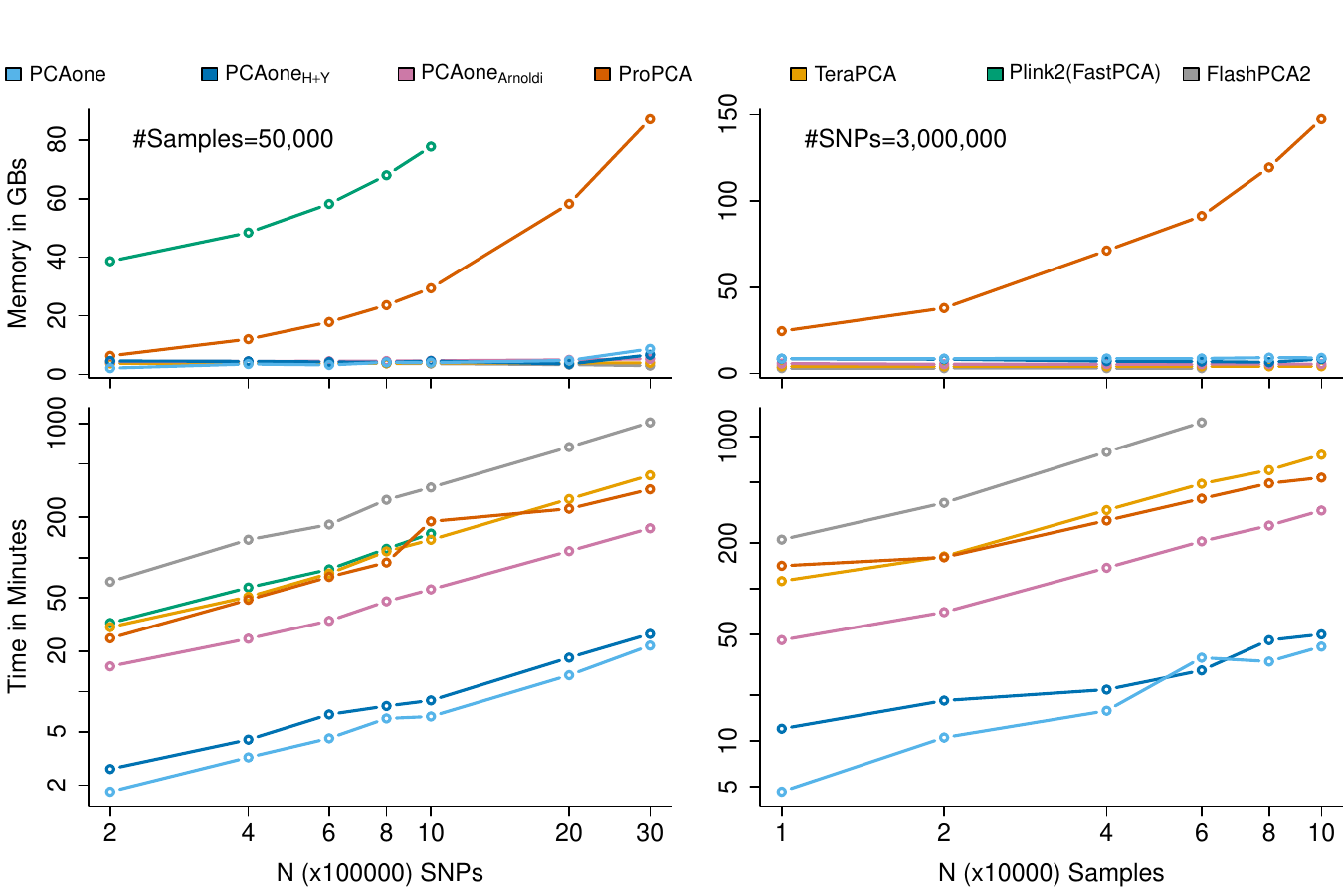
A method for accurate PCA for large scale genetic data. Analyse millions of sites for all 500000 individuals in UKbiobank on your labtop
Zilong Li📨, Jonas Meisner, Anders Albrechtsen📨
HaploNet
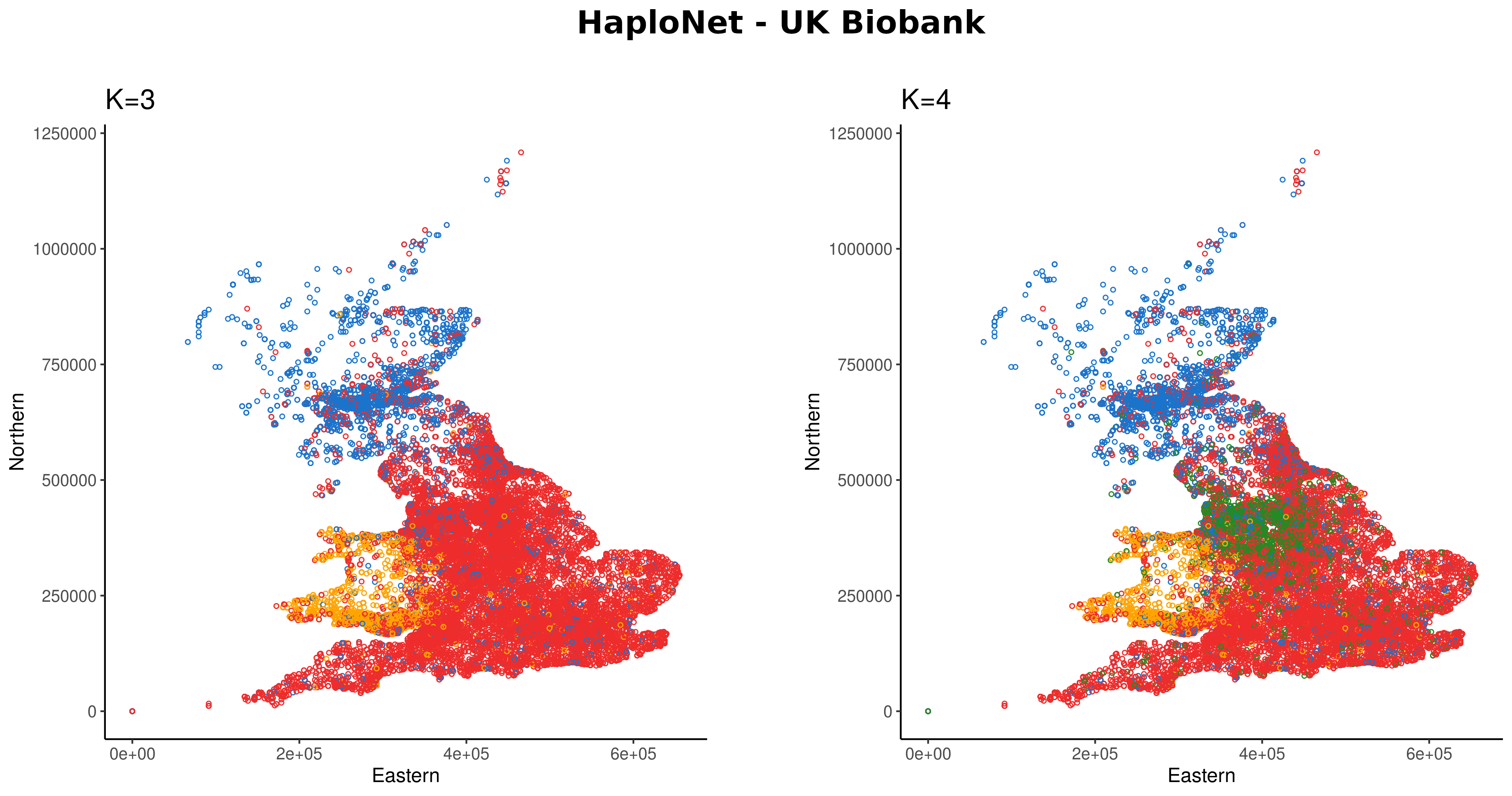
Using sequencing data from simulations and closely related human populations, we show that our approach is better at distinguishing closely related populations than standard admixture and principal component analysis software. We further show that HaploNet is fast and highly scalable by applying it to genotype array data of the UK Biobank. U
Jonas Meisner📨, Anders Albrechtsen
SATC
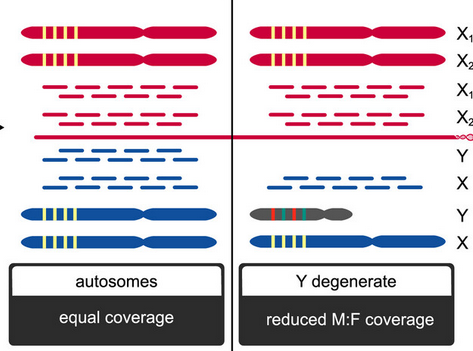
Framework for joint determination of individual sex and sex-linked scaffolds for non-model organism based on depth of coverage
Nursyifa C.* ; Brüniche-Olsen A.* ; Garcia-Erill G.; Heller R.📨; Albrechtsen A.📨
winSFS

Inference of the site frequency spectrum (SFS) from low-depth sequencing data.
Rasmussen M.S.; Garcia-Erill G.; Korneliussen T.S.; Wiuf C.; Albrechtsen A.
EMU
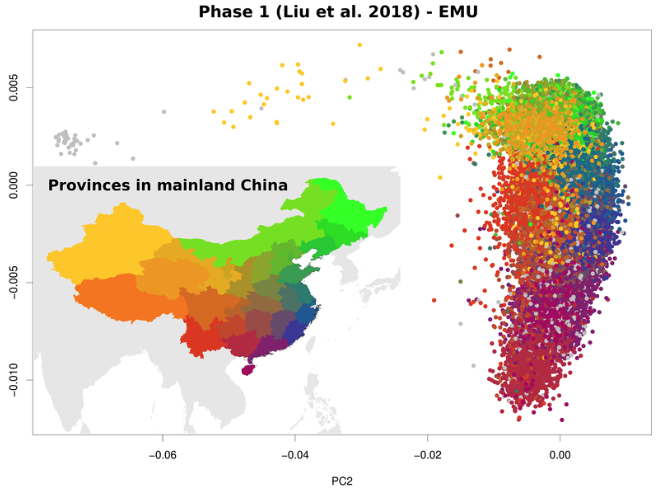
PCA with rampant missingness including having samples with non overlapping data
Meisner J.; Liu S.; Huang M.; Albrechtsen A.
NGSremix

Estimating relatedness coefficients for admixture samples with low depth sequencing. Also works for F1 and other recently admixed indivudals.
Nøhr A.K.; Hanghøj K.; Garcia-Erill G.; Li Z.; Moltke I.; Albrechtsen A.
evalAdmix
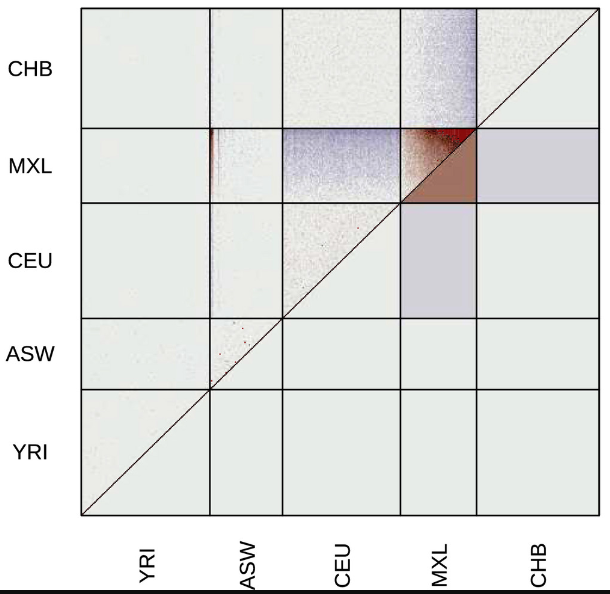
Evaluation your estimated admixture proportions. When running ADMIXTURE or NGSadmix you can evaluate the results.
Garcia-Erill G.; Albrechtsen A.
ASAmap
Genetic association of ancestry specific effects when you do not have information about local ancestry.
Skotte L.; Jørsboe E.; Korneliussen T.S.; Moltke I.; Albrechtsen A.
IBSrelate
KING, RO, R1, statistics for relatedness based only two individuals. No reference panel or allele frequencies needed
Waples R.K.; Albrechtsen A.; Moltke I.
PCAngsd
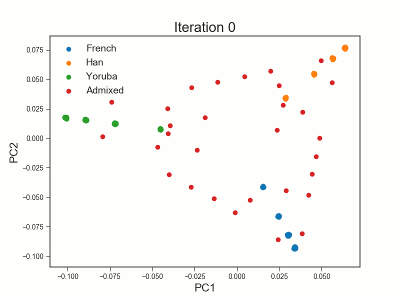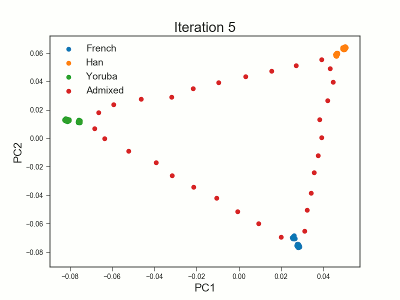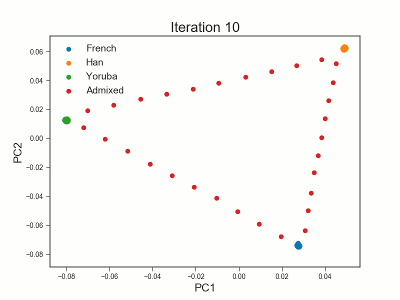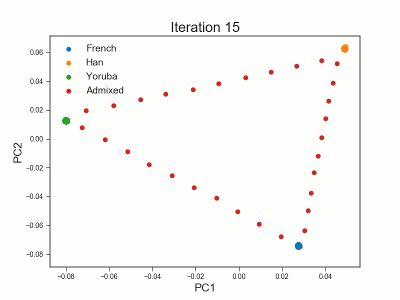
PCAngsd: PCA, admixture proportions, HWE or selection scans from low depth sequencings data while accomidating population structure
Meisner J.; Albrechtsen A.
fastNGSadmix
Jørsboe E.; Hanghøj K.; Albrechtsen A.
ANGSD
Software for population genetic and medical genetic analysis of low depth sequencing data.
Korneliussen T.S.; Albrechtsen A.; Nielsen R.
NGSadmix
Skotte L.; Korneliussen T.S.; Albrechtsen A.
relate
Albrechtsen A.; Korneliussen T.S.; Moltke I.; van Overseem Hansen T.; Nielsen F.C.; Nielsen R.