Research
The Albrechtsen Lab develops statistical and computational methods for genomic data and applies them to questions in medical, population and evolutionary genetics. A recurring theme is how to extract reliable information from large or complex datasets when genotypes, ancestry, relatedness or haplotypes are uncertain.
Our research combines method development with collaborative studies of human populations and non-model organisms. The methods are released as open-source software whenever possible. See the publications and software pages for the full lists.
Multi-omics and molecular mechanisms
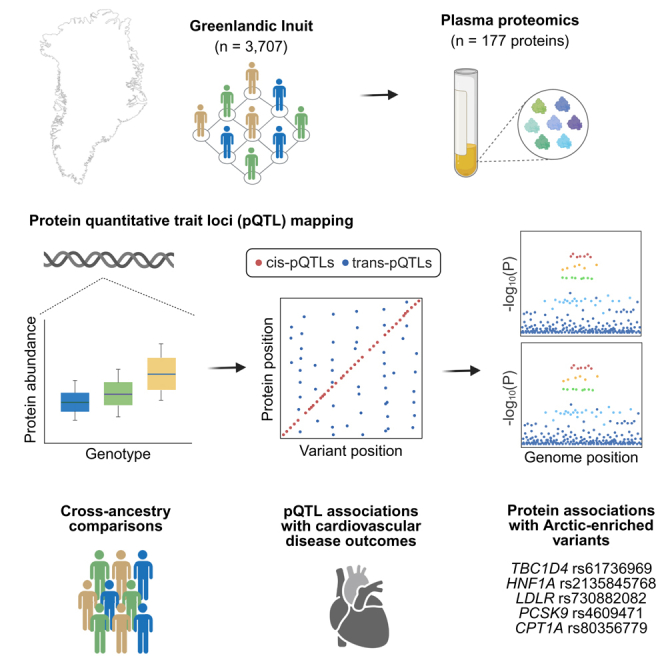
Large genetic studies have identified many variants associated with disease and quantitative traits, but an association alone rarely explains the underlying biology. We integrate whole-genome data with detailed phenotypes and molecular measurements such as transcriptomics, proteomics, metabolomics and microbiome data. The goal is to connect genetic variation to molecular pathways and, ultimately, to disease mechanisms.
Recent work on the Greenlandic plasma proteome combined genetic and proteomic data from 3,707 individuals. The study identified 251 primary protein quantitative trait loci, including 70 previously unreported associations, and linked Arctic-enriched variants to cardiometabolic disease. These datasets also create methodological challenges involving relatedness, admixture, multiple molecular layers and population-specific variation.
Low-depth and read-aware sequencing
Sequencing more individuals at lower depth can be more informative than sequencing fewer individuals deeply, but the resulting genotypes are uncertain. Our methods therefore work directly with sequencing reads or genotype likelihoods instead of relying only on hard genotype calls.
This research includes methods for estimating allele frequencies, population structure, admixture, relatedness, site-frequency spectra and association statistics from low-depth data. QUILT2 performs scalable, read-aware genotype imputation using biobank-sized reference panels and supports short reads, long reads, linked reads, ancient DNA and cell-free DNA. Other tools developed by the group include ANGSD, PCAngsd, NGSadmix and winSFS.
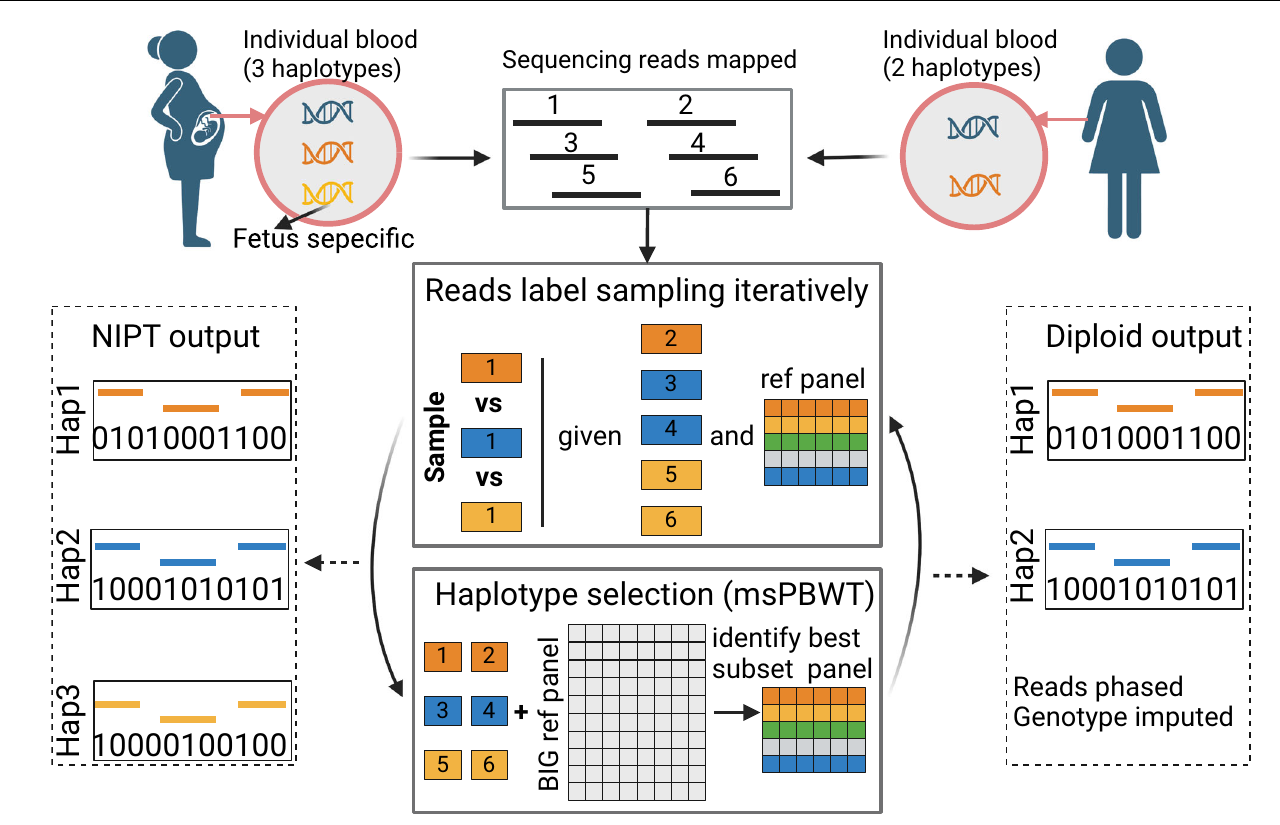
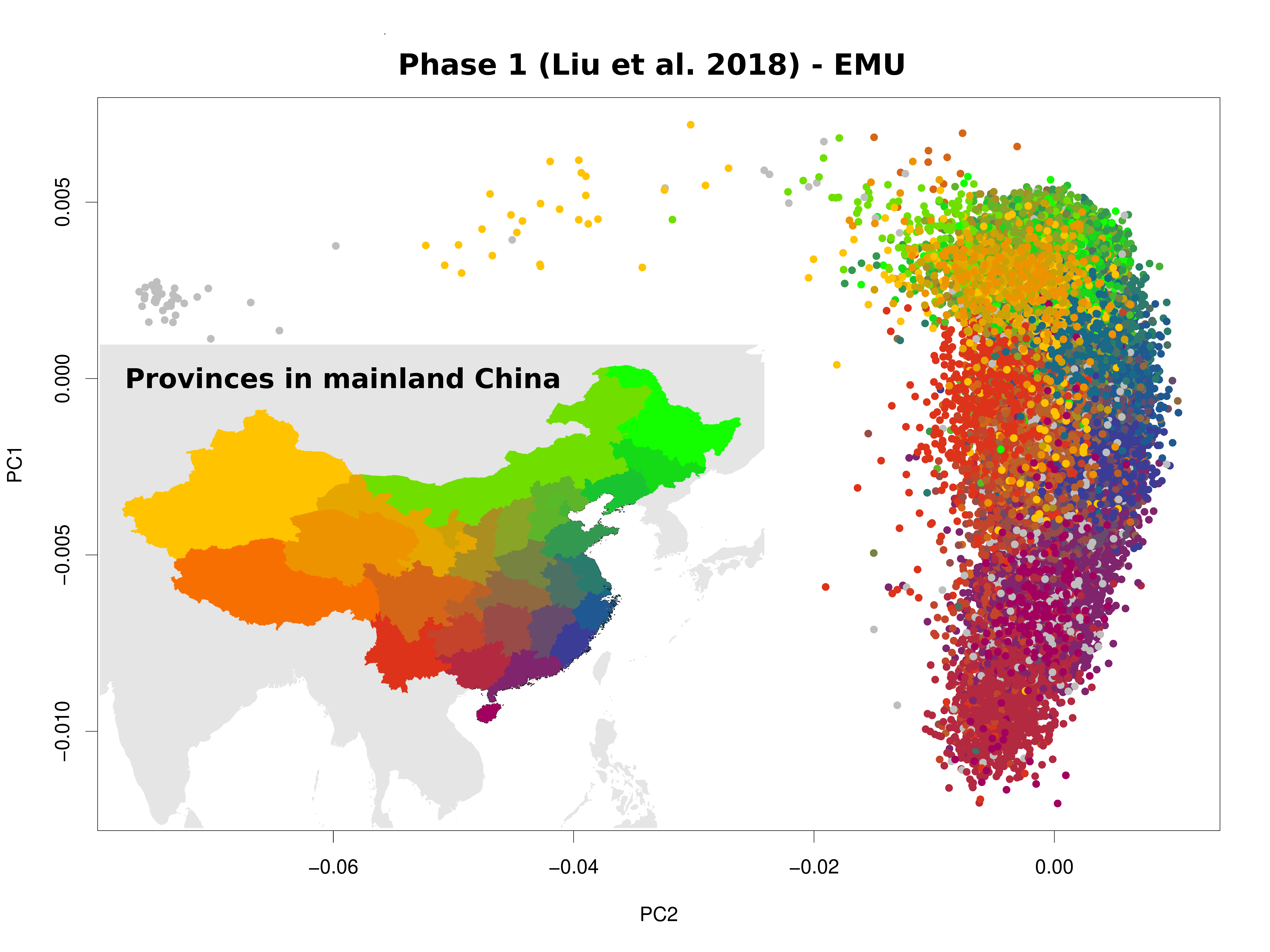
Ultra-low-depth sequencing and NIPT
Non-invasive prenatal testing (NIPT) has generated sequencing data from millions of people, often at less than 0.1-fold genomic coverage. These data have substantial missingness and cannot be analysed reliably with many methods designed for array genotypes or high-coverage genomes.
We develop scalable approaches that account for this uncertainty. The EMU method estimates population structure in data with extensive non-random missingness and was applied to approximately 100,000 individuals from the Chinese Millionome Project. QUILT2 extends this work by jointly modelling reads and reference haplotypes, including a mode designed to impute maternal and fetal genomes from cell-free NIPT data.
Genetics in Greenland
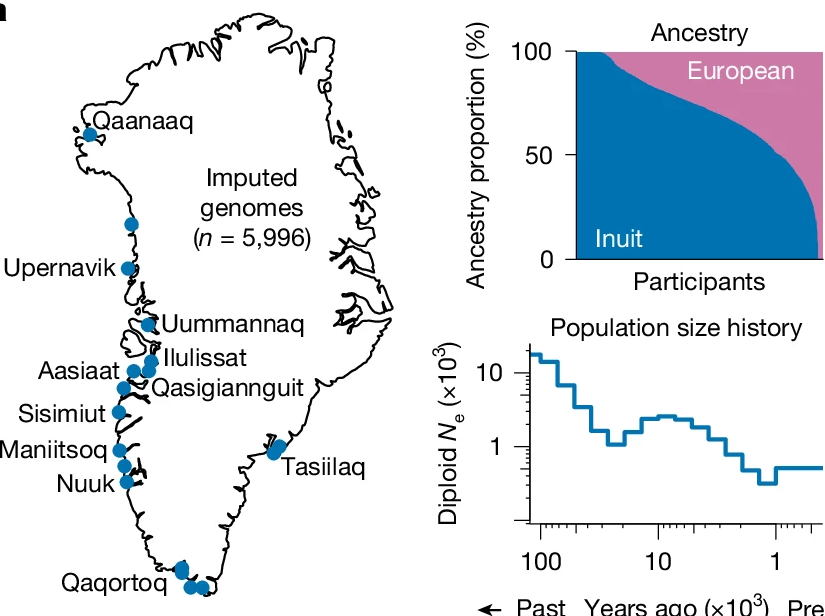
We collaborate on population and medical genetic studies in Greenland. The Greenlandic population has a distinctive demographic history and recent European admixture. Genetic variants that are rare elsewhere can reach high frequencies in Greenland and have substantial effects on health at the population level.
Our work investigates demographic history, natural selection, population structure and the genetic basis of cardiometabolic disease. Studies have identified high-impact variants affecting type 2 diabetes, cholesterol, body weight and other metabolic traits. Recent whole-genome analyses have expanded the catalogue of Greenlandic variation and demonstrated why including underrepresented populations is essential for equitable genetic research and precision medicine.
Population-genetic methods
Population structure, admixture and relatedness can create both biological insight and statistical bias. We develop methods to estimate these quantities, test whether fitted models adequately describe the data and correct downstream analyses when their assumptions are violated.
Current topics include scalable principal component analysis, ancestry-aware linkage disequilibrium, pruning and clumping, admixture-model evaluation, haplotype inference, identity-by-descent and structural-variant genotyping. Representative software includes PCAone, evalAdmix, evalPCA, HaploNet and SVUPP.
The same approaches are used in studies of human history, wildlife conservation and non-model organisms, where sample quality, small population sizes and incomplete reference resources make uncertainty-aware analysis especially important.